![medicamentos]()
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS), como agencia estatal adscrita al Ministerio de Sanidad, Servicios Sociales e Igualdad es el organismo que garantiza a los ciudadanos y a los profesionales sanitarios la calidad, seguridad, eficacia y correcta información de los medicamentos y productos sanitarios que se comercializan en España.
La AEMPS protege la salud pública a través de la autorización previa, el registro y control de la fabricación y comercialización de los medicamentos de uso humano, los medicamentos veterinarios, los productos sanitarios, los cosméticos y productos de cuidado personal y el apoyo a la investigación clínica.
Cómo se evalúan y autorizan los medicamentos
Los medicamentos están regulados a lo largo de todo su ciclo de vida. todos los medicamentos utilizados en España, deben contar con una autorización de comercialización que concede la AEMPS una vez ha evaluado favorablemente su calidad, seguridad y eficacia, y cualquier variación que se produzca debe igualmente ser autorizada o notificada a la AEMPS. Estas evaluaciones permiten asegurar que se mantiene una relación positiva entre el beneficio y el riesgo del medicamento a lo largo de todo su ciclo de vida en el mercado.
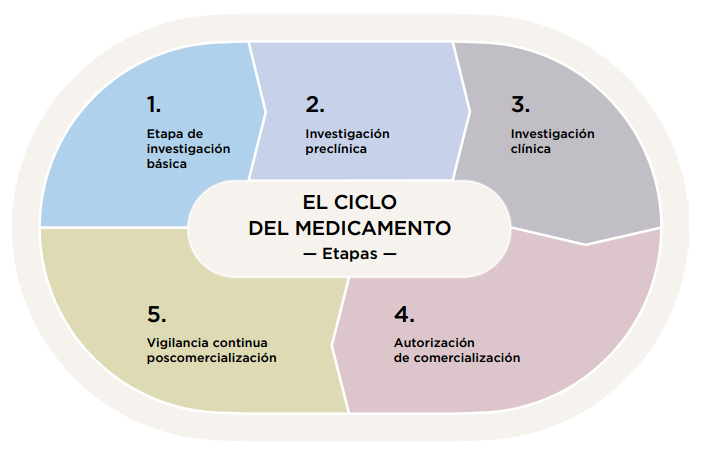
![el ciclo del medicamento]()
Las etapas de investigación de un medicamento
Investigación básica. El descubrimiento de un nuevo medicamento implica procesos como la identificación de candidatos, su síntesis, caracterización, rastreo y pruebas de eficacia terapéutica. A pesar de los avances en tecnología y en conocimiento de los sistemas biológicos se trata de un proceso largo y con una tasa de éxito muy baja. De forma aproximada, se estima que por cada 10.000 moléculas en la etapa de investigación básica sólo 250 entrarán en la siguiente etapa de investigación preclínica
Estudios preclínicos. Tras la investigación básica, las moléculas que resultan más prometedoras son estudiadas en animales de experimentación y en modelos de laboratorio para evaluar su seguridad y actividad biológica. Estos estudios pretenden conocer los efectos del medicamento a distintas dosis en diferentes órganos y sistemas, o cómo se va a distribuir o eliminar el medicamento en el organismo. Se hacen estudios químicos y farmacéuticos sobre el compuesto para conocer su estabilidad o pureza, pruebas de fabricación para determinar si será posible fabricarlo a gran escala, y estudios para preparar la formulación adecuada para su administración.
Ensayos clínicos. Los ensayos clínicos son necesarios para conocer si el comportamiento del medicamento en las personas a las que va destinado, en el caso de los medicamentos de uso humano, o en las diferentes especies animales de destino, en el caso de los medicamentos veterinarios, es adecuado y si consigue realmente eficacia en el tratamiento de la enfermedad para la que se dirige con un perfil aceptable de reacciones adversas.
La autorización. Ningún medicamento puede comercializarse en España sin la autorización previa de la AeMPS o de la comisión europea. La autorización de comercialización se concede en base a criterios científicos sobre la calidad, la seguridad y la eficacia del medicamento de que se trate. Estos tres criterios permiten evaluar la relación entre los beneficios y los riesgos del medicamento para las enfermedades y situaciones para las cuales es aprobado.
Procedimientos de autorización
Procedimiento nacional. El solicitante presenta a la AEMPS el expediente con toda la información para la autorización de comercialización del medicamento en España.
Procedimiento descentralizado. El solicitante presenta su solicitud de autorización de forma simultánea en varios países de la unión Europea. Las distintas agencias evalúan el medicamento de forma coordinada, actuando una de ellas como agencia coordinadora o de referencia y, al final del proceso, todas las agencias emiten una autorización idéntica y válida para su territorio de competencia. ProcedImIento de reconocImIento mutuo. Es el que se utiliza cuando un medicamento tiene ya una autorización de comercialización comunitaria.
Procedimiento centralizado. El solicitante opta a una autorización para todos los Estados miembros de la unión Europea al mismo tiempo. En este caso, el proceso administrativo recae sobre la Agencia Europea de Medicamentos y las evaluaciones científicas son asumidas por dos Estados miembros (ponente y co-ponente), que envían sus informes a los demás Estados miembros.
El expediente de autorización
Una vez superadas con éxito las etapas de investigación del medicamento descritas en el punto anterior, para comercializar un medicamento, es preciso solicitar una autorización aportando en un expediente todos los resultados de la investigación sobre el medicamento, los datos sobre su fabricación, un plan de gestión de riesgos y, en general, toda la documentación que demuestra el cumplimiento de todos los requisitos necesarios para su autorización.
La ficha técnica, el prospecto y el informe público de evaluación
A partir de la evaluación de toda la información que existe sobre el medicamento, la AEMPS desarrolla tres documentos destinados a informar sobre su uso: la ficha técnica, el prospecto y el informe público de evaluación.
La entrada Cómo se regulan los medicamentos y productos sanitarios en España aparece primero en FarmacoVigilancia.